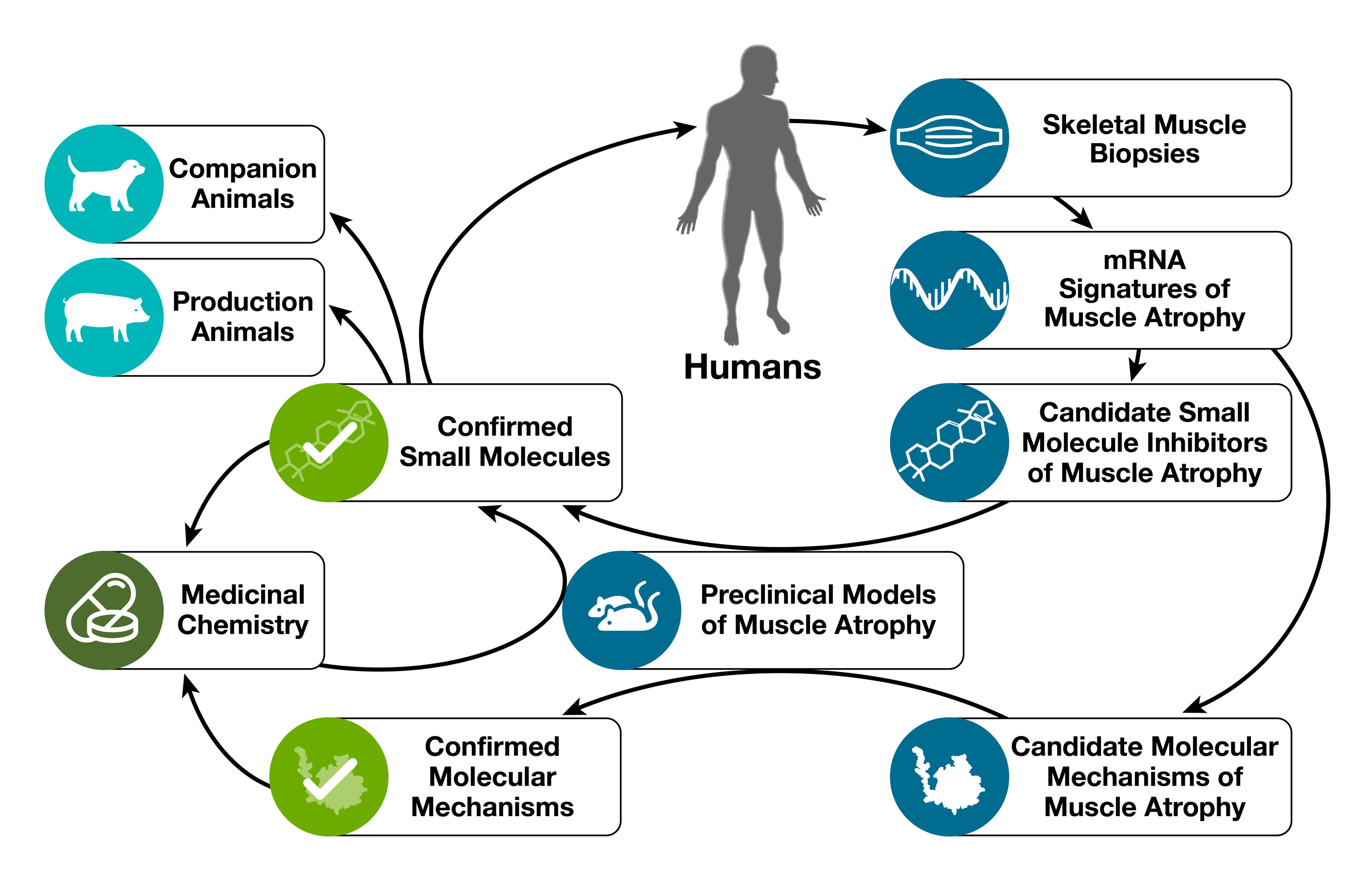
 Our research starts with human skeletal muscle. We obtain muscle biopsies from people with either healthy muscles or muscle atrophy. We use those muscle biopsies to determine mRNA expression signatures of muscle atrophy, which capture all of the gene expression changes that are associated with muscle atrophy in humans. We then search for small molecules whose mRNA expression signatures are opposite to mRNA signatures of human muscle atrophy. Those small molecules represent candidate small molecule inhibitors of skeletal muscle atrophy. For more information on our systems-based discovery process, please see our publication: “Use of mRNA expression signatures to discover small molecule inhibitors of skeletal muscle atrophy”.
Our research starts with human skeletal muscle. We obtain muscle biopsies from people with either healthy muscles or muscle atrophy. We use those muscle biopsies to determine mRNA expression signatures of muscle atrophy, which capture all of the gene expression changes that are associated with muscle atrophy in humans. We then search for small molecules whose mRNA expression signatures are opposite to mRNA signatures of human muscle atrophy. Those small molecules represent candidate small molecule inhibitors of skeletal muscle atrophy. For more information on our systems-based discovery process, please see our publication: “Use of mRNA expression signatures to discover small molecule inhibitors of skeletal muscle atrophy”.
After identifying candidate small molecule inhibitors of muscle atrophy, we perform in vivo studies in mouse muscle atrophy models to confirm or rule out the candidates’ safety and efficacy. Small molecules that demonstrate outstanding safety and efficacy in mouse models (e.g. the natural compounds ursolic acid and tomatidine) are taken forward towards commercial applications in humans, companion animals, and production animals. Some of these molecules are also used as lead compounds in a medicinal chemistry program that generates novel molecules for in vivo testing. Novel molecules that demonstrate improved pharmacologic properties in mouse models of skeletal muscle atrophy are taken forward towards pharmaceutical applications in human patients.
In parallel to our work on small molecules, we use mRNA expression signatures of human muscle atrophy to identify candidate molecular mechanisms of muscle atrophy. We investigate those candidate mechanisms through the use of molecular genetics in mouse muscle atrophy models. Our mechanistic work complements and informs our small molecule work and has allowed us to discover several fundamental molecular mechanisms of muscle atrophy.